
Need a contemporary manufacturing strategy?
Considering the high development costs of medications and the time invested before a drug goes from discovery to market, it is critical to develop not only a solid drug development strategy, but a contemporary manufacturing strategy as well. A parallel development strategy could be one possible approach.
Beginning with in-human Phase I trials, significant thought is applied to study design, logistics, CRO selection, patient populations, study centers, software and clinical distribution. These are all important considerations, but what is often missing is an equally important, well-thought through manufacturing strategy for the clinical material. Choosing a manufacturing partner with the contemporary manufacturing capabilities and expertise to offer advice on future drug product development is critical. The wrong choice can mean biotech companies must switch partners midstream, resulting in high costs and risks, such as failure to produce, the loss of API, or trial delay.
Plan prior to entering first-in-human studies
The best time to involve a manufacturing partner is from the very start. At that time, it is important to have answers to three fundamental questions:
- What are the market requirements for your new molecule, i.e. what is the clear unmet need for your target market, the drugs indication, and the market potential?
- What are the desired product attributes for your drug? For example, what is the clear benefit in the dose scheme/application form?
- What is your commercialisation strategy? Will you license the drug to other companies? If you intend to market it yourself, what markets will you target?
This is why the choice of an experienced manufacturing partner and the development of a clinical manufacturing strategy should be considered prior to entering first-in-human studies.
Proven manufacturing strategies for a biologic or small molecule
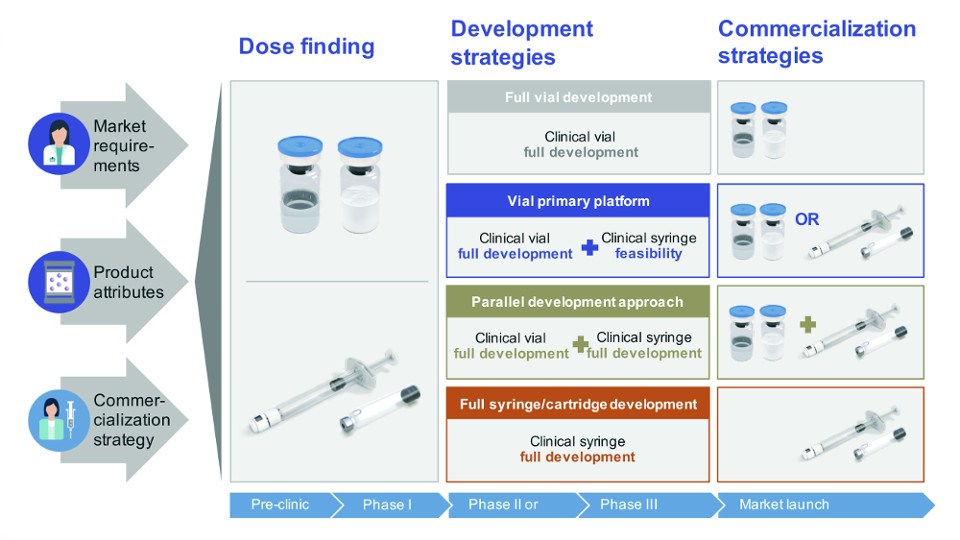
From a CDMO perspective, there are three proven development strategies: full-vial development, parallel-development, and full-syringe or full-cartridge development. Your choice will depend upon your product’s attributes, market requirements, and commercialisation strategy.
The majority of new products are initially filled in vial form since it is easier to carry out dosing studies and the regulatory path is clear. Many biotech companies choose this development path due to their restricted resources, budget, and market-entry strategy. Some exceptions to this rule include accelerated drug development trials or biosimilars. In such cases, the dosing is pre-defined, based on a previous gold standard for a similar compound, and it is known from the start that the final presentation will be a syringe or cartridge.
For a new product or biosimilar where an approved treatment for a patented biologic already exists in syringe or cartridge form, there can be a competitive advantage in using a parallel manufacturing development strategy employing a pre-filled syringe or pen and cartridge combination. This is true in less price sensitive markets where there is a need to attract patients, and vials offer a price advantage. This is also the case when health care workers are paid per administration step, and receive higher reimbursement from insurance companies when using a vial.
The best phase to consider parallel development is following dose-finding studies (Phase IIb). When choosing the parallel development approach, the regulatory pathway can be pursued in parallel. Working with a partner that has strong regulatory experience and is consistently audited to the highest manufacturing quality standards for all clinical phases will help in getting the right answers for submission. Each primary packaging material has its specific regulatory requirements.
In summary, having different packaging presentations is often a choice made when your commercialisation strategy demands a differing price point.
Choosing the right manufacturing partner
There are many local and regional CDMOs and formulation companies with extended small-scale filling services in the market. Many of these companies offer a discount strategy for early phase clinical filling. Their focus is on small-scale preclinical, and early phase filling. This is often sufficient to begin. As soon as your drug product surpasses early clinical phases, however, these partners may have reached their capacities. Thus, you may face issues such as the lack of a plan to limit loss of API in scale up, time delays, and non-cGMP practices, all of which may require additional feasibility, pump and filter studies, and added costs for knowledge and technology transfer to prepare for scale-up.
About Vetter
Headquartered in Ravensburg, Germany, Vetter is a global leading contract development and manufacturing organization (CDMO) with production facilities in Germany and the United States. The company has long-term experience offering services ranging from early development support, including clinical manufacturing, to commercial supply and various packaging solutions for vials, syringes, and cartridges. Vetter’s customers range from small and midsize to the world’s top 20 pharmaceutical and biotech companies. As a leading solution provider, the CDMO recognizes its responsibility to support the needs of its customers in developing devices that contribute to increased patient safety, convenience, and enhanced compliance. Learn more about Vetter at www.vetter-pharma.com.
(First published in European Biotechnology, Autumn Edition 2017)
 adobe stock - aliaksandr
adobe stock - aliaksandr Dr. Dong-Jiunn Jeffery Truong
Dr. Dong-Jiunn Jeffery Truong SLAS - Alexandra Csuport Photography
SLAS - Alexandra Csuport Photography